


Neurodegenerative Disorders
Probing organelle dysfunction at the heart of neurodegenerative disease.

When Organelles Fail: The Subcellular Origins of Neurodegeneration
Neurodegenerative diseases such as Alzheimer’s, Parkinson’s, Huntington’s, and ALS share a common subcellular denominator: the progressive dysfunction of lysosomes, mitochondria, and the endoplasmic reticulum. When these organelles fail to degrade misfolded proteins, sustain energy supply, or maintain calcium homeostasis, neurons degenerate irreversibly.
This article reviews the organelle biology underpinning neurodegeneration, examines TMEM175 as a paradigmatic lysosomal drug target, and explains how ORIA Bioscience’s purified organelle platform opens a direct route to organelle-level drug discovery at scale.
A Global Crisis With a Subcellular Root
Neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS), collectively affect over 50 million people worldwide and are projected to triple in prevalence by 2050 as populations age. Despite decades of research and billions invested in clinical trials, disease-modifying treatments remain limited. The vast majority of candidates have failed in late-stage trials, not for lack of scientific ambition, but because they targeted the wrong layer of biology.
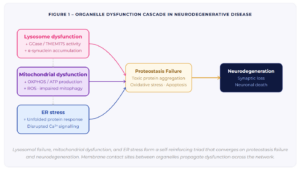
A growing body of evidence now points to a convergent, subcellular explanation: regardless of the disease or the specific genetic risk factor involved, neurodegeneration consistently involves the breakdown of organelle function, primarily in lysosomes, mitochondria, and the endoplasmic reticulum (ER). These are not peripheral bystanders. They are the operational core of neuronal survival, and their dysfunction is increasingly recognised as both a cause and an amplifier of disease progression.

State of the Art: Three Organelles at the Heart of Neuronal Survival
Organelles do not operate in isolation. They form a physically interconnected network that exchanges calcium ions, lipids, and metabolic signals through membrane contact sites (MCSs). Disruption at any node propagates dysfunction across the network. In neurons, which are post-mitotic cells that cannot dilute damage through division, such dysfunction accumulates over decades into irreversible pathology.
Lysosomes: the cell’s degradation hub
Lysosomes maintain an acidic lumen (pH 4.5–5.5) essential for over 60 hydrolytic enzymes that degrade misfolded proteins, damaged organelles, and metabolic by-products via the autophagy-lysosome pathway (ALP). In neurodegenerative diseases, lysosomal acidification failure, impaired enzyme activity, and defective vesicular trafficking converge to prevent the clearance of toxic aggregates, including alpha-synuclein (PD), amyloid-beta and tau (AD), and mutant huntingtin (HD). This proteostatic failure is now recognised as a unifying mechanism across the major neurodegenerative diseases.
Mitochondria: the neuron’s power plant
Mitochondria sustain neuronal survival through oxidative phosphorylation, calcium homeostasis, and regulation of apoptosis. Mitochondrial dysfunction in neurodegeneration manifests as decreased ATP production, elevated reactive oxygen species (ROS), loss of mitochondrial membrane potential, and impaired mitophagy, which is the selective autophagy of damaged mitochondria. Dysfunctional mitochondria that escape clearance become destructive, activating the intrinsic cell death pathway. Given that neurons cannot compensate through glycolysis, mitochondrial bioenergetic failure represents a direct route to neuronal death.
Endoplasmic reticulum: proteostasis checkpoint
The ER is the entry point of the secretory pathway and a major calcium reservoir. In neurodegenerative conditions, accumulation of misfolded proteins triggers the unfolded protein response (UPR), which, when chronic, drives neuroinflammation and apoptosis. ER-lysosome and ER-mitochondria contact sites are particularly sensitive to disruptions in protein trafficking and calcium homeostasis, meaning ER stress frequently amplifies downstream organelle dysfunction.
Case Study – TMEM175, a Lysosomal Ion Channel at the Centre of Parkinson’s Disease
Among the most compelling recent developments in Parkinson’s disease genetics is the identification of TMEM175 (transmembrane protein 175) as a lysosomal ion channel whose dysfunction directly links organelle biophysics to neurodegeneration. TMEM175 resides in the lysosomal membrane, where it mediates K⁺ and H⁺ conductance to stabilise luminal pH, a function essential for the activity of hydrolytic enzymes including glucocerebrosidase (GCase). Loss of this buffering function destabilises lysosomal pH, impairs enzyme activity, and ultimately prevents the clearance of alpha-synuclein aggregates, a hallmark of Parkinson’s disease pathology.
TMEM175 is encoded at one of the most significant loci in Parkinson’s disease GWAS (rs34311866, p = 1.47 × 10⁻⁵⁰, OR = 1.23). The causal variant, p.M393T, reduces lysosomal K⁺ current by approximately 40% and has been directly linked to accelerated disease progression and earlier age of onset. Its structural novelty relative to canonical K⁺ channels, combined with the identification of first-in-class selective inhibitors in 2024, makes TMEM175 one of the most tractable emerging targets in neurodegeneration.
Characterising TMEM175 pharmacology requires direct access to native lysosomal membranes, which whole-cell assays and crude lysates fundamentally cannot provide. Whole-cell patch-clamp cannot resolve lysosomal currents without artefactual vacuolin-based enlargement; cell-based pH assays conflate lysosomal, cytoplasmic, and ER signals. This is precisely where ORIA’s platform becomes enabling.
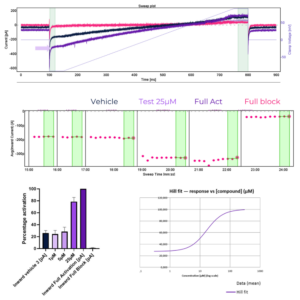
Figure 2 – TMEM175 channel activity in LYSO-Prep™ preparations: inward K+ currents: test compound at 25µM / Normalized currents and corresponding Hill fit
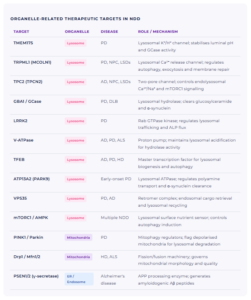
Key Organelle-Linked Targets in Neurodegeneration
The table below lists validated and emerging therapeutic targets in neurodegeneration that are directly linked to organelle dysfunction, with the associated diseases and organelle compartment for each.
Frequently Asked Questions
Neurons are post-mitotic, long-lived cells that cannot dilute accumulated damage through cell division. They rely almost exclusively on mitochondrial oxidative phosphorylation for energy production, unlike most other cell types which can compensate through glycolysis. They also accumulate misfolded proteins over decades, making lysosomal degradation capacity a rate-limiting survival factor. Any disruption to these systems therefore has disproportionate consequences in neurons compared to dividing tissues.
TMEM175 is a lysosomal K⁺/H⁺ channel that stabilises luminal pH by balancing V-ATPase proton pumping. It is encoded at the top Parkinson’s disease GWAS locus (rs34311866, p = 1.47 × 10⁻⁹, OR = 1.23). The causal p.M393T variant reduces K⁺ current by approximately 40%, destabilises lysosomal pH, impairs GCase activity, and increases alpha-synuclein accumulation. Its structural uniqueness relative to canonical K⁺ channels and the 2024 identification of first-in-class selective inhibitors make it one of the most tractable emerging drug targets in neurodegeneration.
LYSO-Prep™ provides greater than 90% pure, functionally intact lysosomes compatible with HTS platforms. For TMEM175 specifically, it enables native-membrane patch-clamp electrophysiology (avoiding artefactual vacuolin-based protocols), direct lysosomal pH profiling, downstream GCase activity measurement, and inhibitor dose-response screening, all at organelle-level resolution not achievable in whole-cell or crude lysate systems.
The landscape of organelle-linked NDD targets is rich. For Parkinson’s disease: TMEM175, GBA1/GCase, LRRK2, PINK1/Parkin, ATP13A2. For Alzheimer’s disease: V-ATPase, PSEN1/2, TPC2, VPS35, TFEB. For ALS and Huntington’s disease: Drp1/Mfn1-2, mTORC1/AMPK, TRPML1. Many targets, including TFEB, V-ATPase, and mTORC1, are relevant across multiple diseases, reflecting the shared organelle dysfunction that underpins the entire NDD spectrum.
Whole-cell assays conflate signals from multiple compartments, including lysosomal, cytoplasmic, mitochondrial, and nuclear, making it impossible to attribute a pharmacological response to a specific organelle. Crude lysates rapidly lose membrane integrity and enzyme activity, eliminating functional channel and enzyme measurements. Purified organelle preparations allow direct biochemical interrogation of a single compartment with intact membranes, native enzyme activity, and physiological ion gradients, producing cleaner SAR data earlier in the drug discovery pipeline.